
In-house medical devices are exempted from most of the provisions of (EU) 2017/745 (Medical Devices Regulation) and (EU) 2017/746 (In Vitro Diagnostic Medical Devices Regulation), given that the health institution follows the conditions laid out in Article 5(5) of the relevant Regulation.
Article 5(5) contains several rules pertaining to the manufacturing and use of in-house medical devices, and the MDCG published its first Guidance of 2023 to provide direction on the application of these rules. This topic pertains to healthcare professionals and researchers of health institutions aiming to design, manufacture, modify, and use in-house devices.
Article 5(5) of the MDR has been fully applicable since 26 May 2021, whereas Article 5(5) of the IVDR is on a scheduled rollout ranging from 2022 to 2028. Refer to Annex B of the Guidance for a detailed timeline.
Medical devices manufactured and used within the same EU health institution on a non-industrial scale, to address specific needs of target patient groups that cannot be met by an equivalent CE-marked device, are known as “In-House Medical Devices.” These devices are exempt from most of the provisions of MDR and IVDR, provided the health institution adheres to the conditions laid out in Article 5(5) of the relevant Regulation.
Article 5(5) defines an in-house device as a device that must be manufactured and used only within the same health institution. Manufacturing a device by a health institution can include creating a device from raw materials, combining a device with another device or product, or modifying an existing device. The use of an in-house manufactured device is considered to be within the health institution when the device is used in the care or diagnosis of a patient. The use of CE-marked devices by healthcare professionals outside the manufacturer’s intended purpose may be subject to national provisions.
Examples of “In-House Devices” include:
- Polymerase Chain Reaction (PCR) master mix used to run PCRs on human DNA/RNA specimens.
- Medical device software, developed by the health institution, used only on-site.
Examples that are NOT “In-House Devices” include:
- Device applications used by the patient outside of the institution to enter data communicated to the healthcare professional.
- Orthopaedic Braces that can be adapted by the patient outside of the health institution.
- Self-tests that are used outside of the health institution.
- Self-Interests (i.e., institution-developed devices that are not clinically relevant).
Refer to section 3.2.3 in the Guidance for further details on what constitutes in-house devices.
IN-HOUSE QMS REQUIREMENTS
The QMS (Quality Management System) for in-house medical devices, including IVDs, should comply with Article 5(5), the relevant requirements of Annex I, national legislation, and applicable ISO standard(s) if the health institution is certified/accredited.
The QMS can cover the whole health institution or only the parts involved in the device’s manufacturing, modification, and use. It should include a process for obtaining information about equivalent CE-marked devices that become available on the market. Table 1, below, provides examples of areas covered in an appropriate QMS.
Table 1 – QMS elements for Health Institution compliance
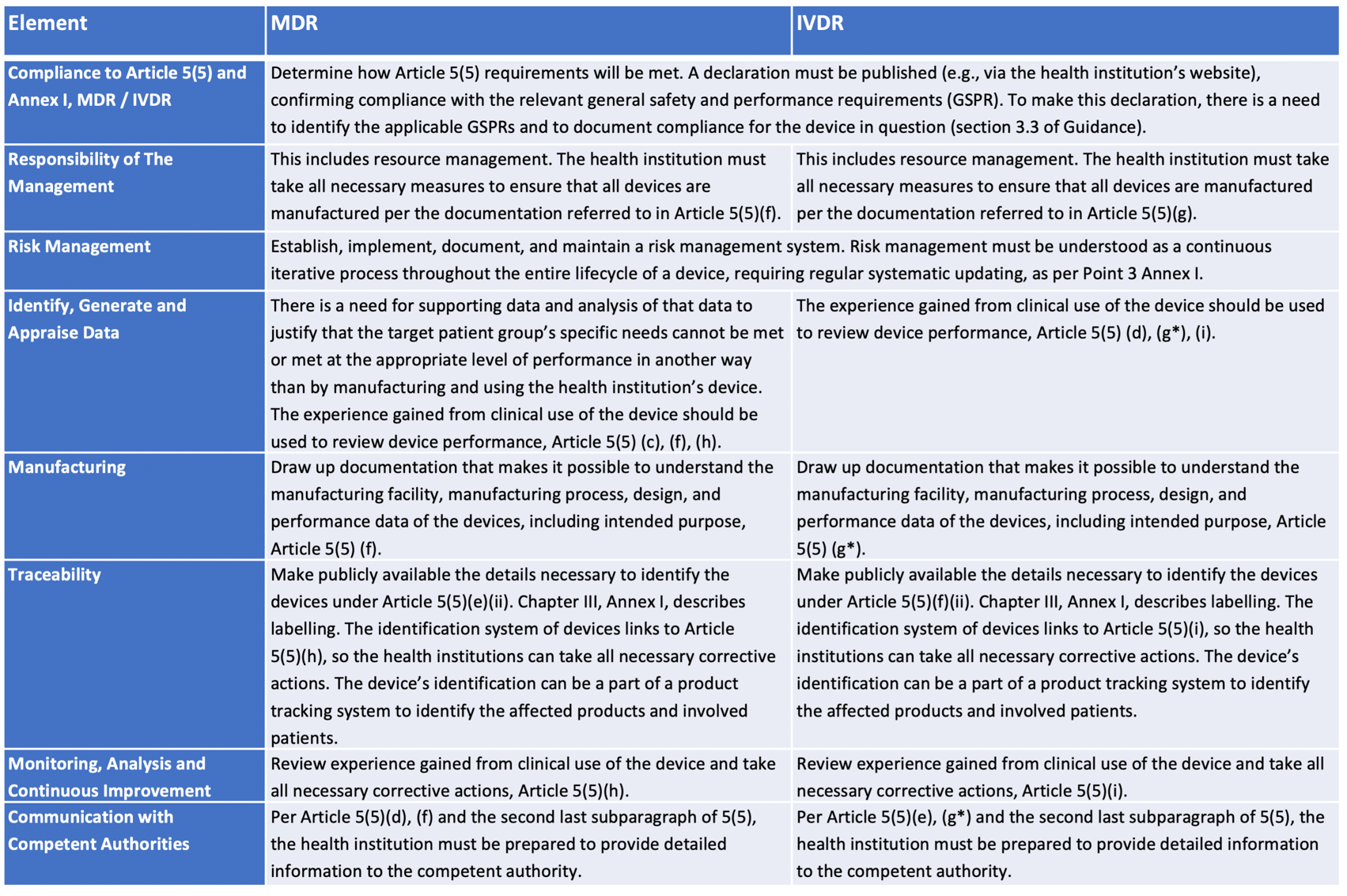
*Applies only to class D devices unless regulated otherwise by national provisions.
GENERAL FACTORS FOR COMPLIANCE
Health institutions must ensure that their in-house manufactured devices comply with the relevant general safety and performance requirements (GSPR, Annex I) of the MDR and IVDR. This includes:
- Establishing a Risk Management System.
- Regularly updating the benefit-risk ratio assessment.
- Ensuring that the device design, manufacture, and performance comply with the relevant provisions.
Health institutions must also ensure that the information supplied with the device complies with the relevant provisions of the MDR and IVDR and regularly update the proof of compliance of their in-house devices.
In the case of In-Vitro Diagnostic Devices (IVDs), the health institution’s laboratory must comply with standard EN ISO 15189 or with national provisions, including accreditation and certification.
EU health institutions should examine the market for the presence and availability of equivalent CE-marked devices before manufacturing an in-house device for the first time. They must then justify and document that the target patient group’s specific needs cannot be met, or met at the appropriate level of performance, by an equivalent device available on the market. This justification should be based on technical, biological, or clinical aspects of the device, such as different intended purposes, clinical conditions, or population groups. In-house devices cannot be transferred to another legal entity and must be manufactured and used under an appropriate quality management system.
Have questions or want to learn more? Visit www.medicept.com or email us at mediceptsales@medicept.com.
Kristina Poberezhnaya – Associate Medical Device Consultant